Forwarded from PhDFinder
📢 Germany – Postdoc in Molecular AI & Quantum Mechanics at Bayer
🏛 University: Bayer AG
🌍 Country: Germany
🎓 Fields:
computational chemistry, materials science, physics, computer science, cheminformatics
🔗 Apply Now: (If the link doesn’t open, tap and hold it, then choose “Open in browser.”)
https://phdfinder.com/2026/02/14/germany-postdoc-in-molecular-ai-quantum-mechanics-at-bayer/
🏛 University: Bayer AG
🌍 Country: Germany
🎓 Fields:
computational chemistry, materials science, physics, computer science, cheminformatics
🔗 Apply Now: (If the link doesn’t open, tap and hold it, then choose “Open in browser.”)
https://phdfinder.com/2026/02/14/germany-postdoc-in-molecular-ai-quantum-mechanics-at-bayer/
PhDFinder
Germany – Postdoc in Molecular AI & Quantum Mechanics at Bayer - PhDFinder
University: Bayer AG
👍4🔥1
ORNL_AISD-Ex: Quantum chemical prediction of UV/Vis absorption spectra for over 10 million organic molecules
Oak Ridge National Laboratory has released unprecedented UV–vis spectral datasets for over 10 million organic molecules, substantially enriching reference data for benchmarking electronic-structure methods and excited-state protocols beyond standard small test sets.
https://doi.ccs.ornl.gov/dataset/13423cfb-df80-541c-a3d9-a2f042fbe507
Oak Ridge National Laboratory has released unprecedented UV–vis spectral datasets for over 10 million organic molecules, substantially enriching reference data for benchmarking electronic-structure methods and excited-state protocols beyond standard small test sets.
https://doi.ccs.ornl.gov/dataset/13423cfb-df80-541c-a3d9-a2f042fbe507
❤9
OpenAlex
All the world's research, connected and open.
Inspired by the Library of Alexandria, we catalog 474 million scholarly works, linking them to authors, institutions, funders, and more—all fully open.
https://openalex.org
All the world's research, connected and open.
Inspired by the Library of Alexandria, we catalog 474 million scholarly works, linking them to authors, institutions, funders, and more—all fully open.
https://openalex.org
❤10
ioChem-BD 4.0 is released (free plan available)
ioChem-BD is the solution to the many problems encountered in performing computational chemistry and materials science discovery research projects. The platform helps you to manage the data produced, analyse the output files and understand the results of your research, and eventually to publish your datasets. The ioChem-BD platform is made to be the daily workspace for your group research activities.
In addition to its main features, ioChem-BD has introduced new developments that incorporate improvements in computational chemistry data management and introduce tools for team members' data administration. These enhancements are part of the premium+ version, offering an advanced solution for managing and analyzing your research data more effectively.
https://www.iochem-bd.com
ioChem-BD is the solution to the many problems encountered in performing computational chemistry and materials science discovery research projects. The platform helps you to manage the data produced, analyse the output files and understand the results of your research, and eventually to publish your datasets. The ioChem-BD platform is made to be the daily workspace for your group research activities.
In addition to its main features, ioChem-BD has introduced new developments that incorporate improvements in computational chemistry data management and introduce tools for team members' data administration. These enhancements are part of the premium+ version, offering an advanced solution for managing and analyzing your research data more effectively.
https://www.iochem-bd.com
❤3🤔1
Fun Fact of the Day
Today is Ludwig Boltzmann birthday (born 20 Feb 1844).
Boltzmann’s constant k_B is what turns “counting microstates” into measurable thermodynamics; in practice, it’s also what makes β = 1/(k_B T) the natural knob controlling the balance between energy minimization and entropy in statistical ensembles, exactly the tradeoff you implicitly navigate when interpreting free-energy surfaces from simulation.
Today is Ludwig Boltzmann birthday (born 20 Feb 1844).
Boltzmann’s constant k_B is what turns “counting microstates” into measurable thermodynamics; in practice, it’s also what makes β = 1/(k_B T) the natural knob controlling the balance between energy minimization and entropy in statistical ensembles, exactly the tradeoff you implicitly navigate when interpreting free-energy surfaces from simulation.
❤9
Forwarded from PhDFinder
📢 France – PhD in Molecular Dynamics at University of Tours
🏛 University: University of Tours
🌍 Country: France
🎓 Fields:
Physics, Chemistry, Materials Science, Nanotechnology, Computational Science
🔗 Apply Now: (If the link doesn’t open, tap and hold it, then choose “Open in browser.”)
https://phdfinder.com/2026/02/17/france-phd-in-molecular-dynamics-at-university-of-tours/
🏛 University: University of Tours
🌍 Country: France
🎓 Fields:
Physics, Chemistry, Materials Science, Nanotechnology, Computational Science
🔗 Apply Now: (If the link doesn’t open, tap and hold it, then choose “Open in browser.”)
https://phdfinder.com/2026/02/17/france-phd-in-molecular-dynamics-at-university-of-tours/
PhDFinder
France – PhD in Molecular Dynamics at University of Tours - PhDFinder
University: University of Tours
👍2
QMCPACK v4.2.0
QMCPACK is an open-source, high-performance electronic structure code that implements numerous Quantum Monte Carlo (QMC) algorithms. Its main applications are electronic structure calculations of molecular, periodic 2D, and periodic 3D solid-state systems. Real-space variational Monte Carlo (VMC), diffusion Monte Carlo (DMC), and a number of other advanced QMC algorithms are implemented. A full set of orbital-space auxiliary-field QMC (AFQMC) methods is also implemented.
https://www.qmcpack.org
QMCPACK is an open-source, high-performance electronic structure code that implements numerous Quantum Monte Carlo (QMC) algorithms. Its main applications are electronic structure calculations of molecular, periodic 2D, and periodic 3D solid-state systems. Real-space variational Monte Carlo (VMC), diffusion Monte Carlo (DMC), and a number of other advanced QMC algorithms are implemented. A full set of orbital-space auxiliary-field QMC (AFQMC) methods is also implemented.
https://www.qmcpack.org
❤5😍2
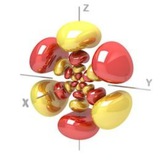
Koopmans’ Theorem
Explanation:
Within the Hartree–Fock (HF) approximation, Koopmans’ theorem states that the first ionization potential (IP) approximates the negative of the highest occupied molecular orbital energy:
IP ≈ −ε_HOMO
This holds under the frozen orbital approximation, i.e., no orbital relaxation upon electron removal.
Key Implications:
• Provides a simple connection between orbital energies and experimental ionization energies.
• Widely used for qualitative assessments of frontier orbital energetics in molecular systems.
Common Misconceptions:
• Not exact: It neglects orbital relaxation and electron correlation; corrections (e.g., via ΔSCF or correlated methods) are often necessary for quantitative accuracy.
• In DFT: There is no formal justification for Koopmans’ theorem with approximate exchange–correlation functionals, although ε_HOMO sometimes empirically correlates with experimental IPs.
Explanation:
Within the Hartree–Fock (HF) approximation, Koopmans’ theorem states that the first ionization potential (IP) approximates the negative of the highest occupied molecular orbital energy:
IP ≈ −ε_HOMO
This holds under the frozen orbital approximation, i.e., no orbital relaxation upon electron removal.
Key Implications:
• Provides a simple connection between orbital energies and experimental ionization energies.
• Widely used for qualitative assessments of frontier orbital energetics in molecular systems.
Common Misconceptions:
• Not exact: It neglects orbital relaxation and electron correlation; corrections (e.g., via ΔSCF or correlated methods) are often necessary for quantitative accuracy.
• In DFT: There is no formal justification for Koopmans’ theorem with approximate exchange–correlation functionals, although ε_HOMO sometimes empirically correlates with experimental IPs.
❤11🤔2
"Constructive atomistic modeling
of matter - 2026"
May 8-10, 2026
Constructor University, Bremen, Germany
Conference covers a broad range of topics, including:
- Quantum simulations of small molecules and bulk systems;
-Classical molecular dynamics
Machine learning approaches for materials property prediction.
The conference aims to bring together a wide spectrum of modern computational techniques and showcase their applications in the simulation of matter. We warmly invite contribution talks from young scientists and look forward to welcoming participants to the cozy campus of Constructor University in Bremen.
Abstract submission deadline: Mar. 27, 2026
Participation is free of charge.
More information here:
https://constructive-atomistic-modeling.de/
of matter - 2026"
May 8-10, 2026
Constructor University, Bremen, Germany
Conference covers a broad range of topics, including:
- Quantum simulations of small molecules and bulk systems;
-Classical molecular dynamics
Machine learning approaches for materials property prediction.
The conference aims to bring together a wide spectrum of modern computational techniques and showcase their applications in the simulation of matter. We warmly invite contribution talks from young scientists and look forward to welcoming participants to the cozy campus of Constructor University in Bremen.
Abstract submission deadline: Mar. 27, 2026
Participation is free of charge.
More information here:
https://constructive-atomistic-modeling.de/
❤7👍3🔥2
Visual Interactive Analysis of Molecular Dynamics v0.1.39 released
VIAMD is an interactive analysis tool for molecular dynamics (MD) written in C/C++. VIAMD is developed at the PDC Center for High Performance Computing (KTH, Stockholm). It exposes a rudementary noscript language that is used to declare operations which are performed over the frames of the trajectory. The results can then be viewed in the different windows exposed in the application.
https://github.com/scanberg/viamd
VIAMD is an interactive analysis tool for molecular dynamics (MD) written in C/C++. VIAMD is developed at the PDC Center for High Performance Computing (KTH, Stockholm). It exposes a rudementary noscript language that is used to declare operations which are performed over the frames of the trajectory. The results can then be viewed in the different windows exposed in the application.
https://github.com/scanberg/viamd
❤8